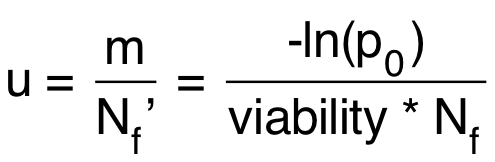Determination of Mutation Rate by Fluctuation Analysis
Adapted by Seung-Been Steven Lee from the Saccharomyces cerevisiae protocol of Greg Lang and based on the original Luria-Delbruck bacterial protocol. Last updated: November 2013.
References
Luria, S.E., and Delbruck, M. (1943). Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics 28, 491-511.
William A. Rosche and Patricia L. Foster. Determining Mutation Rates in Bacterial Populations. Methods (2000) 20, 4-17 .
Determination of Mutation Rate using Fluctuation Assay. Greg Lang (personal communication).
Overview
 Among many different ways to determine mutation rate, fluctuation analysis—especially with the p0 method—is useful because it is relatively easy and quick. First described by Luria and Delbruck (1943) for use in a bacterial system, the p0 method allows one to calculate mutation rates with relatively simple procedures. Unlike other methods, it does not involve computer simulations, complex mathematical theories, or calculus. The purpose of this protocol is to provide a detailed outline of the method along with the parameter optimizations needed for a successful assay.
Among many different ways to determine mutation rate, fluctuation analysis—especially with the p0 method—is useful because it is relatively easy and quick. First described by Luria and Delbruck (1943) for use in a bacterial system, the p0 method allows one to calculate mutation rates with relatively simple procedures. Unlike other methods, it does not involve computer simulations, complex mathematical theories, or calculus. The purpose of this protocol is to provide a detailed outline of the method along with the parameter optimizations needed for a successful assay.
The basic idea of the fluctuation analysis is to start growing many replicate cultures of cells that initially have no mutation of interest, to let them go through a certain number of cell divisions to saturation—so as to limit the number of generations—in a permissive medium, and to measure how many of the cultures have not acquired the mutations. There are two successive selections: the first one selects for absence of the mutation while the second one selects for presence of the mutation. The first selection ensures that the starting population doesn’t already contain cells with the mutation—if the starting cells already contain the mutations, it would be impossible to determine accurately the rate at which the mutation is occurring within the cells. The cells are then grown in permissive condition for a defined window of time during which mutations may accumulate. By subsequently selecting only those cells that have acquired the mutations, it is easy to exclude cells that have failed to obtain the mutation—those cells cannot form colonies. When growing multiple replicate cultures, the permissive medium must provide conditions where the mutation is neither selected for nor selected against (i.e., the mutation should not have a deleterious effect in that medium). The number of cell divisions that occurred in each replicate culture can be estimated by measuring the final cell population of the culture. Knowing the total number of cell divisions and the number of cultures that failed to give colonies in the second selection (i.e., in which there had been no mutation), one can then calculate the mutation rate.
The p0 method (the Poisson distribution) requires that the proportion (p0) of cultures without mutants should be between 10% and 80%. Therefore, it is critical to identify optimal culture conditions and thus the optimal culture size that will give rise to the appropriate number of cellular generations at saturation (so as to yield a fraction of zero-mutation events that is in the desired range). There are numerous ways to control the number of cell divisions that occur in a culture by the time it reaches saturation:
- Sugar concentration (e.g., 2% vs. 0.1% vs. 0.05%)
- Culture volume (e.g., 100 µL vs. 50 µL vs. 30 µL)
- Initial number of cells in the culture, N0 (e.g., 100 cells vs. 500 cells vs. 1,000 cells)
Many different combinations of these factors can be used to get the correct size of zero-class events. Because the mutation rate can differ between mutation types, strains, and experimental conditions, you may have to repeat this optimization step several times. However, once you have figured out suitable conditions so that p0 is between 10% and 80% for a particular strain, you can use those conditions for that particular strain throughout your experiment. Four different conditions that were tested for use in an actual experiment are shown below; only the dextrose concentration was varied:
- 0.1% Dextrose, 30 µL Complete medium, N0 =1000 cells
- 0.05% Dextrose, 30 µL Complete medium, N0 =1000 cells
- 0.01% Dextrose, 30 µL Complete medium, N0 =1000 cells
- 0.005% Dextrose, 30 µL Complete medium, N0 =1000 cells
1. |
Grow an overnight culture of the strain to be tested in medium that selects against growth of cells in which the mutation has already occurred. For example, if you are comparing chromosome loss rates, you have to use selective media that will guarantee the presence of the test/control chromosome before introducing cells into permissive medium in step 3 below. You can test whether or not your selective medium is sensitive enough (i.e., it sufficiently prevents phenotypic lagging) by plating those cells grown in the overnight culture directly onto plates that select for the mutation (e.g., loss of the chromosome). If you find that the culture already yields colonies on the plates, you have to cut down the initial cell number (N0) and repeat the process. If changing N0 does not work, you either have to introduce an additional selection into your strain or find other methods to determine the mutation rate (for alternative methods, see William A. Rosche and Patricia L. Foster. Determining Mutation Rates in Bacterial Populations. Methods (2000) vol. 20: pp 4-17). |
2. |
Sonicate the culture to break apart cell clumps. |
3. |
Determine the cell density of the overnight culture as precisely as possible. You can use a flow-cytometer, a hemocytometer, a spectrophotometer (to measure optical density), etc., but once you choose a method, be consistent with how you count your cells throughout the experiment. |
| 4. | Based on the cell density obtained from step 3, dilute the overnight culture into a large volume (e.g., 30 mL) of permissive medium (complete medium in this case) with four different Dextrose concentrations (0.1%, 0.05%, 0.01%, 0.005%). Now each 30 mL solution should contain approximately 1000 cells per 30 µL or 1,000,000 cells in total. |
| 5. | Confirm your dilutions by measuring cell densities of the four conditions. |
| 6. | Into two 96-well plates, divide samples into wells (48 wells per condition), 30 µL each. Mark regions of wells for each different glucose concentration. |
| 7. | Cover your plates with sealing films, otherwise medium in the cultures will evaporate and the cells may not grow as many generations as expected. You have to periodically exchange gases that are built up in wells by switching the sealing films; usually once a day is enough. This presumably helps to prevent anaerobic growth. |
| 8. | Incubate both plates at the desired temperature for your strain (e.g., 23 °C for ts strains) without shaking for cultures to reach saturation. |
9. |
After the saturation time determined by the method described in step 8: Obtain cell densities in 10 out of 48 wells for each condition and make sure that cell counts vary as expected with the dextrose concentrations. Sonicate before counting. For the rest of the wells, add 70 µL of autoclaved glass distilled (agd) water to the 30 µL cultures so that there is enough volume to plate (100 µL). Mix the contents of the wells before spotting because cells on the bottom of each well will not readily come out. Place the entire volume of each culture onto a dried selective plate. To dry the plates, place a sterilized Whatman filter paper disk that was cut to fit the plate directly on the agar surface. After the Whatman paper is completely soaked (about 2-3 min), remove and discard it using sterile forceps. The plate should now have decreased moisture content—the quicker the cultures are absorbed, the more evenly the cells will be distributed within the spot. |
10. |
Incubate the selective plates for a pre-determined amount of time at the right temperature for your strain until visible and countable colonies appear. The appropriate incubation time can be estimated by growing cells on the selective plate and seeing how long it takes those cells to form visible and distinctive colonies. Alternatively you can micro manipulate single cells and determine how long they take to form visible colonies. |
11. |
Calculate the fraction of zero-class events. Once you find conditions that give an appropriate fraction of zero-class events (10%-80%), you are ready for the actual fluctuation test. If there are too many (more than 80%) or too few (less than 10%) zero-class events, repeat this whole optimization step again to adjust the number of zero-class events. For example, if you observed too many mutations for all dextrose concentrations (you should still see that the densities of mutants within each spot on the plates should decrease along with the sugar concentration), you can try cutting down the volume. Using the lowest dextrose concentration 0.005% Dextrose, test 10 µL, 15 µL, 20 µL, and 25 µL. |
Fluctuation assay
1. |
Grow an overnight culture of the strain to be tested in medium that selects against growth of cells with the mutation. |
2. |
Sonicate. |
3. |
Measure the cell density of the overnight culture. |
4. |
Dilute the overnight culture into a large volume of permissive medium as determined in “Optimization of culture size.” |
5. |
Confirm your dilution but this time by testing for viability instead of obtaining cell density. To do this test, directly dispense from the large volume mixture an appropriate number of cells for counting colonies on permissive plates. The count of colony-forming units will be later used as the initial cell number for each well, which is useful for calculating how many generations of growth have occurred. This test will also account for any cell misbehavior, measurement error, dilution error, etc. |
6. |
Divide the sample into four 96-well plates using the culture volume determined in the “Optimization of culture size.” |
7. |
Cover two of the four plates right away with seal films. Incubate those plates at the right temperature for your strain without shaking and allow the cultures to reach saturation as determined in step 7 of “Optimization of culture size.” |
8. |
Place the entire volume of each culture in the remaining two plates onto a plate that selects for the mutation, using the dried plate technique described above (if using small volume cultures, bring volume up to 100 µL before plating). This initial selection ensures that the cells in each well do not have any mutation at the outset. However, should you observe a few spots that contain mutations from this test, you can still mask the effect of those outliers by subtracting that number from the number of spots in the final selection test. For example, if you observe growth in 3 out of 182 spots in this initial selection (prior to non-selective growth) and 50 out of 182 in the final (following non-selective growth), you can conclude that in reality 47 out of 179 cultures acquired the mutation during the period of non-selective growth. In addition, to avoid any bias coming from subjective calling of “a colony,” set a colony size threshold below which you just ignore growth even if you can see it (e.g., 0.5 mm diameter). |
9. |
After the pre-determined saturation time, measure the final cell density using some of the wells (usually 10 wells are sufficient) from which you can calculate the final cell count Nf. Don’t forget sonication. Use those wells that were counted to also test for viability. Use the average viability obtained from this step for calculation of mutation rate. Place the entire volume of each culture from the plates onto a plate that selects for the mutation, using the dried plate technique described above (again, if using small volume cultures, bring volume up to 100 µL before plating). |
10. |
Incubate the selective plates for a pre-determined amount of time at the appropriate temperature for your strain until colonies are visible and countable. Be consistent with the colony size you determined in step 8. |
11. |
Calculate the mutation rate using the equation below:
u = mutation rate = probability of mutation per cell per division Nf’ = realized Nf (taking account of viability)
Confidence Limits (CLs) can be easily calculated in the statistical software program R using the command:
where P0 = number of zero class cultures and t = total number of cultures (note that P0 is the actual number of zero-class events and is not the same as p0, the fraction of zero-class events). The command returns the +95% and -95% confidence limits for p0 from which the values of m can be calculated. |